WP4: Digital Precision Diagnostics
The DoMore! project aims to develop novel diagnostic and prognostic methods that are easily integrated into standard clinical routine, as well as a method to digitize and automate existing tasks in pathology.
One main aim in the DoMore! project is to develop methods that are easily integrated into standard clinical routine. Our main methods DNA ploidy and Nucleotyping have been developed on specially prepared samples from the cell suspension, called monolayers, where intact cell nuclei are spun onto a glass slide and imaged with a microscope. Both the special preparation and the microscope are factors that decrease the availability of the methods. It is thus an aim to develop methods that work on routine histological sections scanned with a scanner rather than a microscope.
DNA ploidy
Deviations from the normal configuration with two copies of each chromosome are relatively common in cancer cells and are associated with a worse prognosis for the patient. DNA ploidy by image cytometry estimates the total DNA content in cancer cell nuclei and classifies a sample as normal (diploid) or abnormal (non-diploid) based on the evaluation of a histogram of DNA content in about 1500 cell nuclei. The method is well established, and its prognostic impact in several cancer types is well documented [ref Danielsen et al. Nat Rev Clin Oncol. 2016 May;13(5):291-304]. In the DoMore! project we extend the DNA ploidy method first to make use of scanners rather than microscope, and then to be able to analyse the nuclei directly in routine sections, thus eliminating the need for special preparations of monolayers.
DNA ploidy on a scanner
High-resolution scanners will become common equipment in pathology departments with the digitalisation of pathology. Scanning of slides is highly automated in these systems, with functions for scanning a rack of 400 or more slides overnight. We have adapted the DNA ploidy method on monolayers to work on the scanner. Due to the slightly lower resolution on the scanner compared to the microscope, the ability to detect small differences in DNA content is better on the microscope system, but for the majority of non-diploid samples, the DNA content representing the abnormal cell population is significantly higher than the normal cell population and as such not a problem for practical purposes. We have compared the resulting DNA ploidy classification in the two methods in 246 colorectal cancer samples where 236 (96%) had identical classification with the two methods. The few with different classification were due to cell populations with slightly aberrant DNA content compared to the normal diploid cell population where the scanner system did not identify them as aberrant.
The clinical implication of such hyperdiploid subpopolations is unclear and studies are underway to compare the prognostic power of these two different ways of analysing ploidy distributions in tumors.
DNA ploidy in histological sections
Methods that can be applied to routine sections are easier to implement in the clinic. The overall aim of this particular project is to develop a method for the estimation of DNA ploidy status in HE-stained routine sections. An important step in the development process is to estimate DNA ploidy status in Feulgen-stained histological sections; the HE-stain can be removed and replaced with Feulgen-stain and imaged before the section is restained with HE-stain. Feulgen-stain is DNA-specific and stoichiometric and thus allows for more precise measurement of DNA content than the HE-stain. The main challenges in the development of a method for the estimation of DNA content in histological sections are 1) the segmentation of cell nuclei and 2) the DNA content estimation from a profile (5µm) of a cell nucleus. A method for the automatic segmentation of cell nuclei has been developed based on deep learning with convolutional neural networks (see work package 3). Methods for the estimation of DNA content in thin histological sections have been reported earlier (e.g., Haroske et al. 1993) and we have implemented a variant of the method proposed by Haroske et al., and find good correspondence between the monolayer DNA ploidy estimate and the histological section estimate in tissues with near spherical shaped nuclei, such as prostate. In a dataset of 236 samples where we estimated DNA ploidy in both monolayers and histological sections, 188 (80%) had the same classification. So, the preliminary data shows promise, but further work is on-going to optimize and adapt the method.
Nucleotyping
Chromatin structure in cancer cell nuclei is related to transcriptional activity and other cellular processes. We have developed a framework to characterize the chromatin structure based on grey level entropy in Feulgen-stained cell nuclei, where the disorder of grey levels reflects the DNA distribution in local regions. This characterization, termed Nucleotyping, has been developed on monolayer samples and found to discriminate cancer patients with different prognosis. The approach has been validated in datasets of different cancer types. In the DoMore!-project we have demonstrated that Nucleotyping is a general marker across cancer types (Kleppe et al. Lancet Oncol. 2018 Mar;19(3):356-369) and that the proportion of nuclei inhabiting variants of these properties also are strong prognostic markers across gynaecological cancer types (Nielsen et al. J Natl Cancer Inst. 2018 Dec 1;110(12):1400-1408).
We now focus our work on transferring the concept to histological sections, which are different from the specially prepared samples from cell suspension used till now for Nucleotyping. The cell nuclei are not complete due to the sectioning of 5µm sections and require more advanced methods to be automatically segmented. On the other hand, the cells are imaged in their original context in the tissue providing new opportunities for characterization. The previously described method for automated segmentation of cell nuclei in tissue sections with convolutional neural networks is also used in this project. We currently work on the colorectal cancer dataset (about 600 patients treated at Aker University Hospital) in which the automated segmentation method is developed as well as another colorectal cancer dataset (The Gloucester Colorectal Cancer Study with more than 900 patients) to develop the Nucleotyping method in histological sections. Till now we have implemented the methods used for Nucleotyping in monolayers and will evaluate their prognostic impact. Preliminary results are promising with trends indicating a prognostic role for the same methods as in monolayer samples. Extensions of the existing method include incorporating the contextual information from the surrounding tissue as well as the implementation of novel methods for the quantification of chromatin properties.
Automatic Gleason scoring
Donald Gleason was an American physician and pathologist who identified tissue patterns in prostate cancer specimens that were associated with patient prognosis. A grade between 1 and 5 was assigned each of the tissue patterns, such that a higher grade corresponded with more aggressive disease. The dominant and predominant grades were identified, resulting in a Gleason score representing the two main tissue patterns, e.g., 3+4(=7). The Gleason grading system assessed by uropathologists is a very strong prognostic marker for prostate cancer patients and is routinely assessed in this patient group. Specialized pathologists are a scarce resource and increasingly so with the rising incidence numbers for this cancer type.
Furthermore, the intra- and interobserver variation for Gleason scoring is significant. There is thus a need for alternative methods for the assessment of Gleason score in prostate cancer tissue specimens. We have developed a method that estimates the Gleason score in HE-stained tissue sections from prostate cancer specimens based on cell organisation and duct features. The method identifies cell nuclei and ducts, calculates a minimum spanning tree based on the cell nuclei positions and uses features from the minimum spanning tree together with features describing the ducts in a support vector machine classifier to estimate a Gleason grade to the image. The method has been trained and validated on a set of images with homogeneous Gleason grade assessed by uropathologists. The validation results are good, and we are currently working on implementing a version of the application that has a simple user interface, and that can be applied to whole sections.
Automatic mitotic index assessment
The proportion of cells undergoing cell division is a prognostic marker for several cancer types, where a higher proportion for most cancer types is associated with a worse prognosis. The counting of cells undergoing mitosis is one method to assess the degree of cell proliferation in a tissue sample. Pathologists normally count the number of mitotic figures in ten high-power fields, providing a standardized count for the patient. The process is laborious, and the inter- and intraobserver variation is significant. There is thus the interest in an accurate and computerized measurement of this property. We have trained a convolutional neural network to identify mitotic figures in a publicly available breast cancer dataset used in challenges in biomedical imaging such as the Tumor Proliferation Assessment Challenge (TUPAC). The resulting neural network model was then validated on a set of leiomyosarcomas and indicated that the method works as intended. We continue our work with improving the neural network model and extending the validation dataset to comprise all uterine sarcomas in Norway between 1970 and 2000 as described by Abeler et al. (ref Abeler et al. Histopathology. 2009 Feb;54(3):355-64).
When completed, the plan is to validate this method on a large breast cancer material to demonstrate clinical feasibility and implementation.
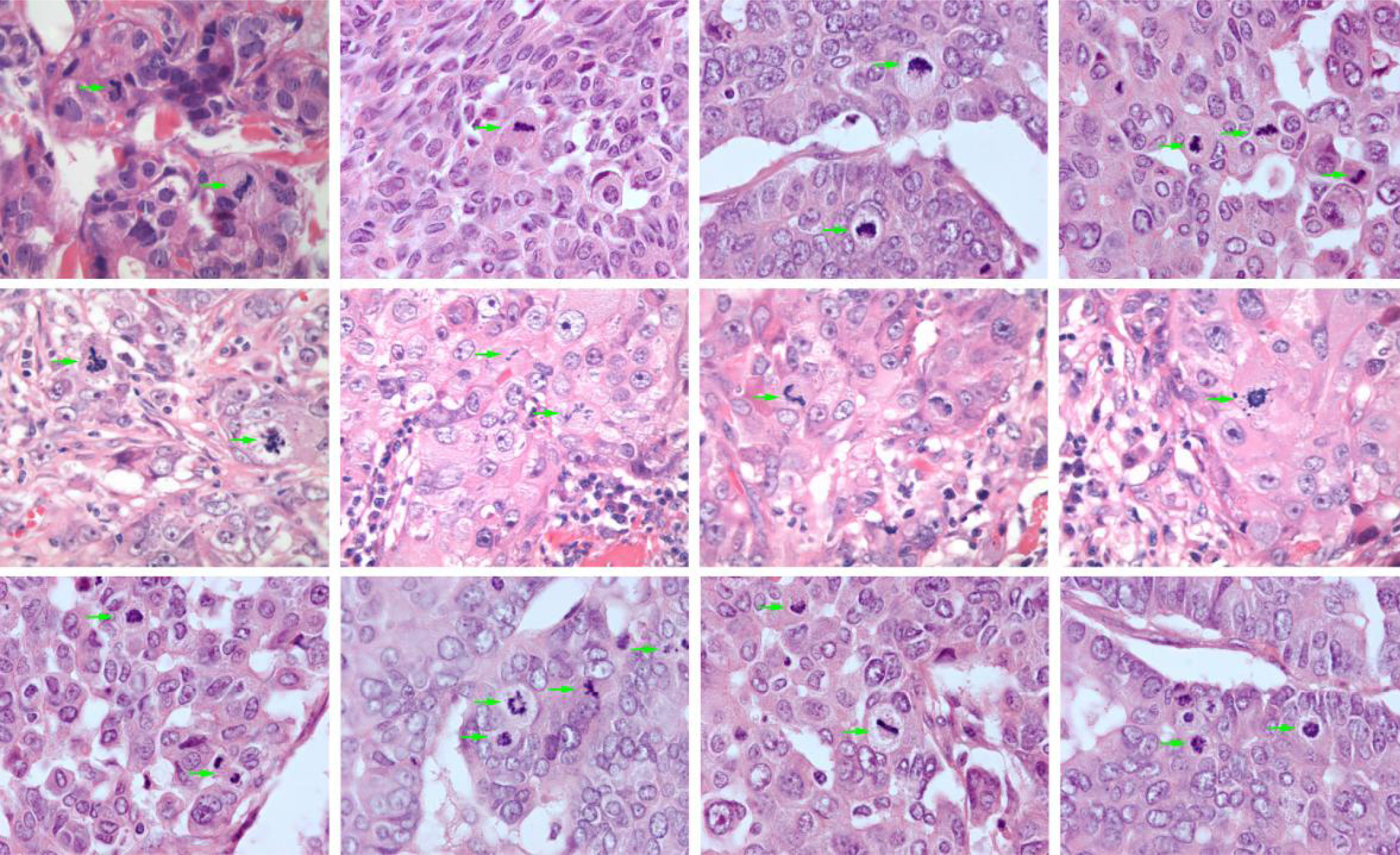 Examples of mitotic index annotation in breast cancer tissue samples.
Examples of mitotic index annotation in breast cancer tissue samples.
Automated estimation of stroma fraction
The tumor microenvironment is the cellular environment in which a tumor exists. A tumor and the surrounding microenvironment are closely related and interact constantly. For simplicity, the tumor is often analyzed in isolation although the dependence on the surrounding tumor environment is well known. Stroma is a major component in the tumor microenvironment, and the stroma-to-epithelial proportion has been reported to be a prognostic factor for colorectal cancer patients based on manual assessment [e.g., Huijbers et al. Ann Oncol. 2013 Jan;24(1):179-85], where a higher stroma fraction is associated with a worse prognosis. We have developed a method to assess the stroma fraction in HE-stained routine histological sections automatically and validated its prognostic impact in colorectal [Danielsen et al., Ann Oncol. 2018 Mar 1;29(3):616-623] and prostate cancer [Ersvær et al., Int J Cancer . 2020 Aug 15;147(4):1228-1234]. We use a fixed threshold to categorize the stroma fraction as low or high. Furthermore, we have combined the stroma fraction estimate with DNA ploidy status to integrate prognostic information from a tumor and its microenvironment. The method can be easily integrated into a clinical routine at a low cost.
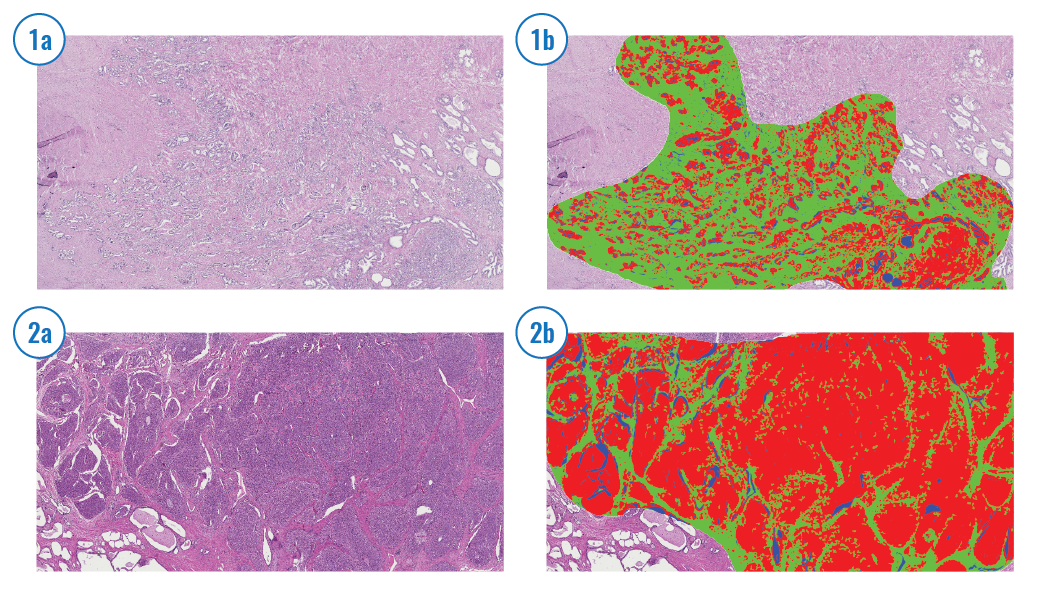 Stroma detection Stroma cells are automatically detected and shown in red.
Stroma detection Stroma cells are automatically detected and shown in red.
Oslo, 15th August 2020
Navigate to the other Work Package descriptions: